Parsing Variants Using the cyvcf2 Library
Parsing Variants Using the cyvcf2 Library
Background
In our previous article on the Variant Call Format (VCF), we described how a human could read and understand the contents of a VCF file. In this article, we are going to describe how a program could. Specifically, a program written in Python using the cyvcf2 library (citation).
Along with this article, is a GitHub repository that includes all of the sample code and VCF files for easier usage. That repository can be cloned here:
$ git clone https://github.com/genomoncology/using-cyvcf2.git
$ cd using-cyvcf2Why cyvcf2
As described in this paper, by library author Brent Pedersen of the Quinlan Lab of the University of Utah, the cyvcf2 parser was created to maximize both performance and approachability for parsing of VCFs.
Below are the relative performance of cyvcf against 3 other popular libraries.
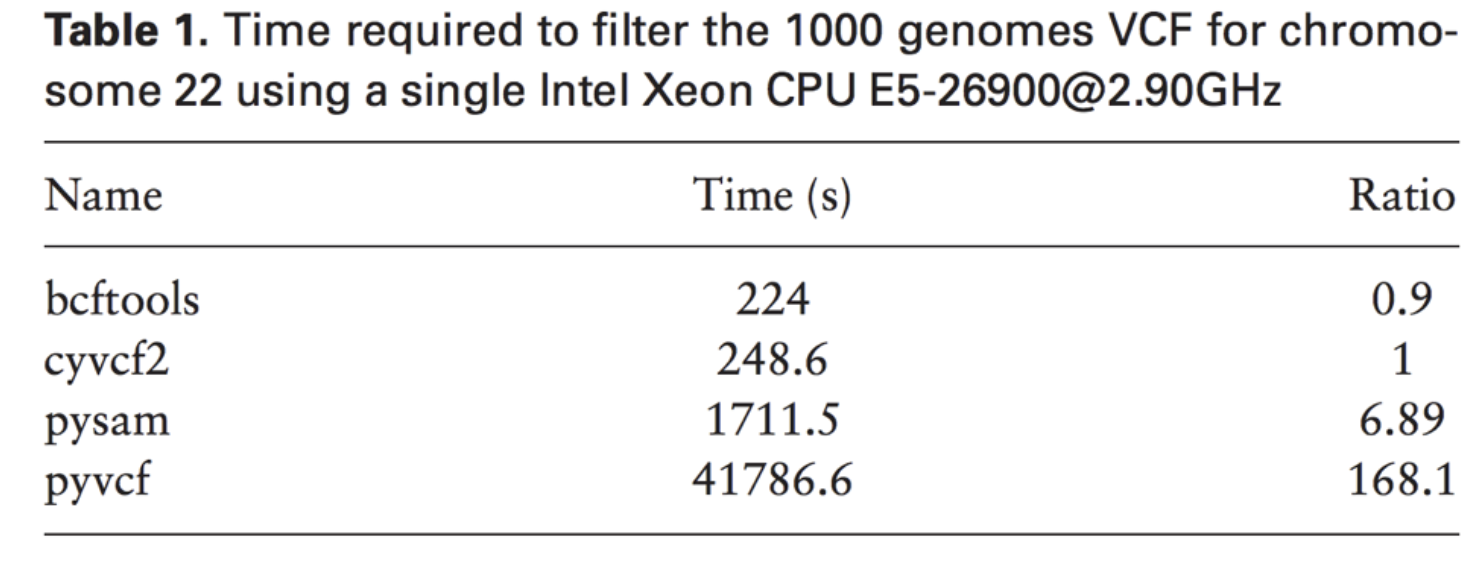
The cyvcf2 library achieves significant performance improvements over the two most popular Python libraries (pysam and pyvcf) and comparable performance to the C-based library bcftools, which is less approachable for most bioinformatics developers. It achieves this performance, by wrapping the high-performance C-based htslib library with Cython, which exposes a Python API.
Installing cyvcf2
This article uses a local installation of python on a Mac using Homebrew and pyenv as described by this tutorial for setting up a local python development environment.
$ python -- version
Python 3.7.5
# create and activate virtualenv
$ python -m venv .venv
$ source .venv/bin/activate
# install library
$ export LDFLAGS="-L/usr/local/opt/openssl/lib"
$ export CPPFLAGS="-I/usr/local/opt/openssl/include"
$ pip install cyvcf2
# check installation
$ pip freeze
click==7.1.2
coloredlogs==14.0
cyvcf2==0.20.0
humanfriendly==8.2
numpy==1.18.4Opening and Iterating a VCF File using VCFReader
We have created a VCF file that matches the one in the VCF specification in our GitHub repository. It is named “spec.vcf”.
It can be used in this first example:
$ python <<EOF
from cyvcf2 import VCF
vcf = VCF("./spec.vcf")
for record in vcf:
print(record)Pasting the above into the command (without the $ prompt) and ending with a CTRL+D will render the following output:

Exploring Cython Objects
Unfortunately, one side effect of using Cython is that there isn’t a __dict__ object and the vars() function doesn’t work, which limits the explorability in the Python REPL.
To help explore, we created a simple function that grabs the interesting key/values from the object and prints a table using the handy tabulate library:
from tabulate import tabulate
def cython_obj_to_dict(obj: object) -> dict:
keys = dir(obj)
keys = filter(lambda k: k[0] != "_" and not
k.isupper(), keys)
data = [(k, getattr(obj, k, None)) for k in keys]
data = [(k, v) for k, v in data if not callable(v)]
data = dict(data)
return data
def display_obj_values(obj: object):
data = cython_obj_to_dict(obj)
print(tabulate(data.items()))This code is available here in our GitHub project in the python module called functions.py and requires the tabulate library to be installed:
$ pip install tabulateUnderstanding a VCF object
Opening a VCF file creates a VCF object, which can be displayed:
>>> from cyvcf2 import VCF
>>> vcf = VCF("./spec.vcf")
>>> vcf
<cyvcf2.cyvcf2.VCF object at 0x11af27e10>
>>> from functions import display_obj_values
>>> display_obj_values(vcf)
-----------------------------------------------------------------
raw_header ##fileformat=VCFv4.2
##FILTER=<ID=PASS,Description="All filters passed">
##fileDate=20090805
##source=myImputationProgramV3.1
##reference=file:///seq/references/1000GenomesPilot-
NCBI36.fasta
##contig=
<ID=20,length=62435964,assembly=B36,species="Homo
sapiens">
##phasing=partial
##INFO=<ID=NS,Number=1,Type=Integer,Description="# of
Samples With Data">
##INFO=
<ID=DP,Number=1,Type=Integer,Description="Total
Depth">
##INFO=<ID=AF,Number=A,Type=Float,Description="Allele
Frequency">
##INFO=
<ID=AA,Number=1,Type=String,Description="Ancestral
Allele">
##INFO=<ID=DB,Number=0,Type=Flag,Description="dbSNP,
build 129">
##INFO=<ID=H2,Number=0,Type=Flag,Description="HapMap2
membership">
##FILTER=<ID=q10,Description="Quality below 10">
##FILTER=<ID=s50,Description="Less than 50% of
samples have data">
##FORMAT=.
<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=
<ID=GQ,Number=1,Type=Integer,Description="Genotype
Quality">
##FORMAT=
<ID=DP,Number=1,Type=Integer,Description="Read
Depth">
##FORMAT=.
<ID=HQ,Number=2,Type=Integer,Description="Haplotype
Quality">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA00001
NA00002 NA00003
samples ['NA00001', 'NA00002', 'NA00003']
seqlens [62435964]
seqnames ['20']
-----------------------------------------------------------------Below is an explanation of each attribute key on the cyvcf2.VCF object:
Understanding a Variant object
The VCF object is an iterator (i.e. it implements the “__iter__” method) so it can be looped through via a for loop.
It can also be explored via the next method:
>>> first_variant = next(vcf)
>>> first_variant
Variant(20:17330 T/A)Calling next, over and over again, will eventually throw a StopIteration exception which is how Python knows how to exit a for-loop:
>>> while True:
... print(repr(next(vcf)))
...
Variant(20:17330 T/A)
Variant(20:1110696 A/G,T)
Variant(20:1230237 T/)
Variant(20:1234567 GTC/G,GTCT)
Variant(20:1234000 G/*)
Traceback (most recent call last):
File "<stdin>", line 2, in <module>
File "cyvcf2/cyvcf2.pyx", line 527, in
cyvcf2.cyvcf2.VCF.__next__
StopIterationTo explore the first variant object, we use display_obj_values function:
>>> from functions import display_obj_values
>>> display_obj_values(first_variant)
------------------ ----------------------------------------
aaf 0.5
call_rate 1.0
end 14370
genotype <cyvcf2.cyvcf2.Genotypes object at
0x113bcb7b0>
genotypes [[0, 0, True], [1, 0, True], [1, 1,
False]]
gt_alt_depths [-1 -1 -1]
gt_alt_freqs [-1. -1. -1.]
gt_bases ['G|G' 'A|G' 'A/A']
gt_depths [-1 -1 -1]
gt_phases [ True True False]
gt_phred_ll_het []
gt_phred_ll_homalt []
gt_phred_ll_homref []
gt_quals [48. 48. 43.]
gt_ref_depths [-1 -1 -1]
gt_types [0 1 3]
is_deletion False
is_indel False
is_snp True
is_sv False
is_transition True
nucl_diversity 0.6
num_called 3
num_het 1
num_hom_alt 1
num_hom_ref 1
num_unknown 0
ploidy 2
start 14369
var_subtype ts
var_type snp
------------------ ----------------------------------------The values of this output have been put into the below table along with corresponding descriptions from the python help() function.
Accessing Info and Format Values
As discussed in our previous article, the VCF file format is a very flexible format that allows for position-level attributes in the INFO section and sample-level attributes described by FORMAT and access for each sample.
To access this information, the Variant object has an INFO dictionary-like attribute and a format() method that accept a key name to retrieve the values:
For example, there is both INFO and FORMAT data named “DP” for Depth in the Specification example:
>>> first_variant.INFO["DP"]
14
>>> first_variant.format("DP")
array([[1],
[8],
[5]], dtype=int32)Calculating Variant Allele Frequency (VAF)
One of the critical data elements to be extracted from VCF files is the Variant Allele Frequency (VAF), which is represented by the above gt_alt_freqs field in cyvcf2.
Unfortunately, not every VCF represents the input values required (reference, alternate and total depths) with the same keys. From reviewing the source code, cyvcf2 supports the formats generated by the GATK and Freebayes variant callers.
In another example, the alt (AO), ref (RO), and total (DP) depths are 2, 47, and 49, respectfully:
#CHROM POS REF ALT QUAL FORMAT 703094
7 140453136 A T 2 GT:AO:RO:DP:GQ
0/1:2:47:49:.Below is how the cyvcf2 library stores these values and calculates VAF % (~4.1%):
gt_alt_depths : [2]
gt_ref_depths : [47]
gt_depths : [49]
gt_alt_freqs : [0.04081633]Determining the VAF of alternate alleles is a critical feature for all of our clients. At GenomOncology, where we integrate with a variety of DNA sequencers and variant callers, we have invested in making our VCF processing software highly configurable to quickly adapt to new VCF formats that we may encounter.
Summary
If you have any questions, please feel free to write to me on twitter (@imaurer) or post an issue on our GitHub repository called “using-cyvcf2”.