What is a Variant Call Format (VCF) file?
Update 2/16/2022
A variant call format file (VCF file) is the output of a bioinformatics pipeline. It specifies the format of a text file used in bioinformatics for storing gene sequence variations. Typically, a DNA sample is sequenced through a next generation sequencing system (NGS system), producing a raw sequence file. That raw sequence data is then aligned, creating BAM/SAM files as a result. From there, variant calling identifies changes to a particular genome as compared to the reference genome. That output is stored in a variant call format, VCF for short.
Watch the video below to learn more about the VCF file, the components of a VCF file, and how to parse a VCF file using Cyvcf2 (a Python library used to parse VCF files).
Below details the links identified in the video.
Background
Variant Call Format (VCF) is a specification [1] for storing genotype data in a tab-delimited file format. Below is a high-level diagram of a typical bioinformatics pipeline that produces a VCF file:

Originally developed for the 1000 Genomes Project [2], the VCF specification has become the de facto standard output for variant calling software due to its concise format and the increase of sequencing data generated from the Next Generation Sequencing (NGS) methods.
File Format
Main Sections
As described in the specification for the Variant Call Format (VCF), there are 3 main sections to each file:
Meta Information Lines - Multiple lines prefixed by double pound symbols (##).
Header Line - Single line prefixed with a one pound symbol (#).
Data Lines - Remainder of the file with 1 position per line.
Meta Section
The Meta section describes the format and content of that specific VCF file. This can include information about the sequencing performed, the variant calling software, or the reference genome used for determining variants. The first few rows from the VCF specification demonstrate this type of information:
##fileformat=VCFv4.3
##fileDate=20090805
##source=myImputationProgramV3.1
##reference=file:///seq/references/1000GenomesPilot-NCBI36.fasta
This Meta section also declares and describes the fields provided at both the site-level (INFO) and sample-level (FORMAT) in the Data Lines. Below are some examples of each type from the VCF specification document:
##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
##INFO=<ID=AF,Number=A,Type=Float,Description="Allele Frequency">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth">
This design allows for great flexibility in the data represented by any given VCF file, allowing each variant calling pipeline to capture the most accurate data and metadata appropriate possible.
However, this flexibility comes at a cost because downstream processing software may need to account for differences in output formats. At GenomOncology, where we integrate with a variety of DNA sequencers and variant callers, we have invested in making our VCF processing software highly configurable to quickly adapt to new VCF formats that we may encounter.
Header Line
Each VCF file has a single header line that has 8 mandatory fields separated by tabs that represent columns for each data line:
#CHROM POS ID REF ALT QUAL FILTER INFO
If there is genotype data, then a FORMAT column is declared and followed by unique sample names. All of these column names must be separated by tabs, as well.
Data Lines
Each data line represents a position in the genome. The data corresponds to the columns specified in the header and must be separated by tabs and ended with a newline.
Below are the columns and their expected values. In all cases, MISSING values should be represented by a dot (‘.’).
#CHROM - Chromosome identifier. Examples include 7, chr7, X or chrX.
POS - Reference position. Sorted numerically in ascending order by chromosome.
ID - Unique identifiers separated by semicolons. No whitespaces allowed.
REF - Reference base (ACGT). Insertions can be represented by a dot.
ALT - Comma-separated Alternate base(s) (ACGT). Deletions represented by a dot.
QUAL - Quality score that is on a log scale. 100 means 1 in 10^10 chance of error.
FILTER - Indicates which filters have failed (semicolon-separated), PASS or MISSING.
INFO - Site-level (non-sample) information in semicolon separated name-value format.
FORMAT - Sample-level field name declarations separated by semicolons.
<SAMPLE DATA> - Sample-level field data separated by semicolons corresponding to FORMAT field declarations.
Example Data Explanation
Specification File
The specification includes an example VCF file.

Position and Ref/Alt Information
Below are some notes to help understand the first 5 columns about the above file.
All of the variants occur on Chromosome 20 on the NCBI36 (hg18).
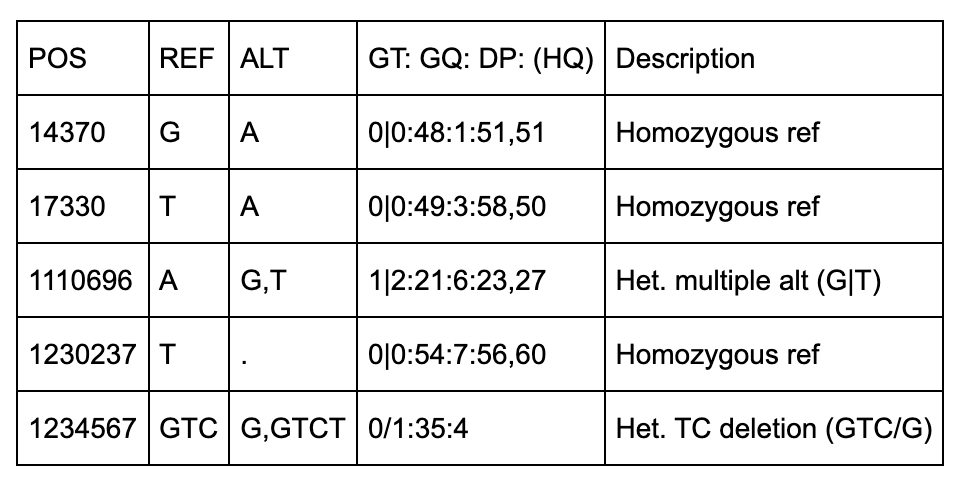
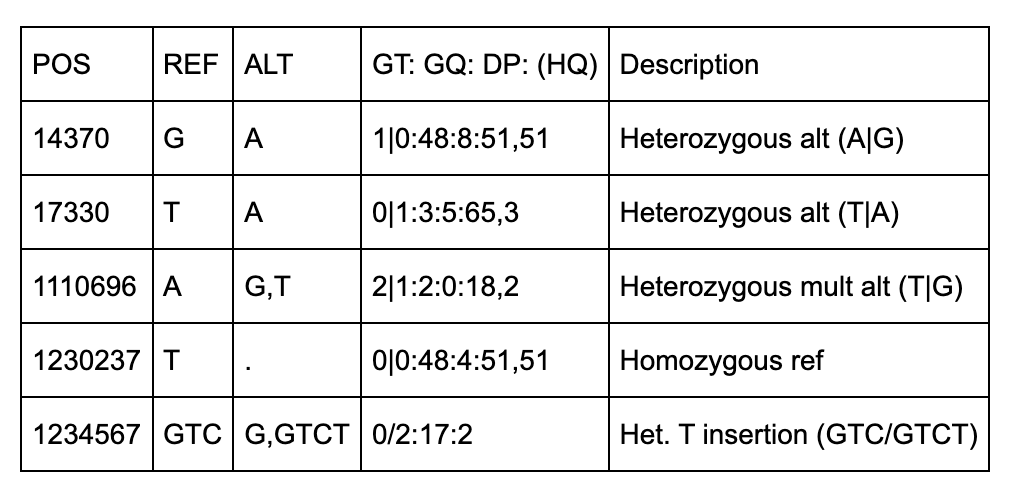
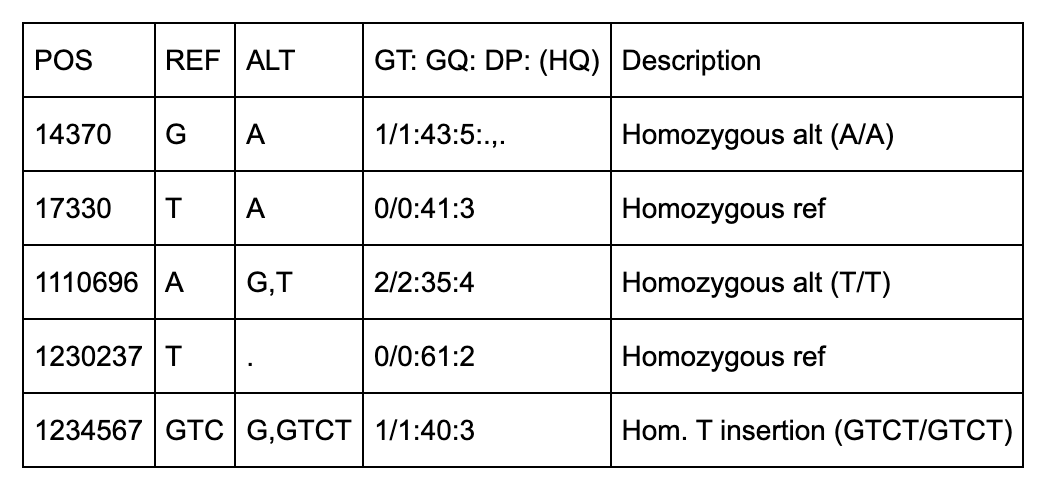
There are 5 positions identified (14370, 18330, 1110696, 1230237, 1234567).
Three of the variants have IDs including 2 dbSNP records (rs6054257, rs6040355).
The first two positions (14370, 17330) are simple single-base pair substitutions.
The third position has 2 alternate alleles specified (G and T) that replace the ref (A).
The fourth position represents a deletion of a T since the alt allele is missing (“.”).
The fifth row has 2 alt alleles, the first is a deletion of TC and second is insertion of a T.
QUAL and FILTER columns
The QUAL column indicates the quality level of the data at that site. The FILTER column designates what filters can be applied. The 2nd row (position 17330), has triggered the q10 filter, which is described in the meta section as “Quality below 10”.
Each bioinformatics pipeline treats these columns differently, so you will need to consult your pipeline’s subject matter experts on how to best interpret this information.
INFO column
The info column includes position-level information for that data row and can be thought as aggregate data that includes all of the sample-level information specified.
FORMAT column
The format column specifies the sample-level fields to expect under each sample. Each row has the same format fields (GT, GQ, DP, and HQ) except for the last row which does not have HQ.
Each of these fields is described in the Meta section as the following:
GT (Genotype) indicates which alleles separated by / (unphased) or | (phased).
GQ is Genotype Quality which is a single integer.
DP is Read Depth which is a single integer.
HQ is Haplotype Quality and has 2 integers separated by a comma.
Sample and Genotype Information
This VCF file has 3 samples identified by their names (NA00001, NA00002, NA00003) in columns 10 through 12. Below are the relevant columns for each of the samples.
NA00001
NA00002
NA00003
References
[1] Variant Call Format Specification
http://samtools.github.io/hts-specs/VCFv4.3.pdf
[2] The variant call format and VCFtools https://academic.oup.com/bioinformatics/article/27/15/2156/402296